Methods to enhance transient transfection of adherent cells have identified many different ligands that improve plasmid uptake into cells. Of these ligands, serum-derived transferrin is well-known to improve transfection efficiency of adherent cells. However, to date, a serum-free and blood-free transferrin has not been tested for improvements to transfection efficiency. We have thus created a protocol for the improvement of transfection efficiency and viral titer without the need for blood-derived transferrin.
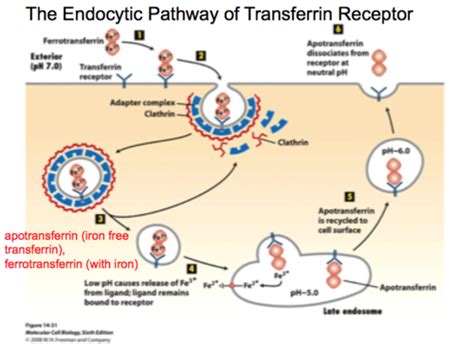
Transient transfection relies on the spontaneous fusion of plasmid DNA with the cell membrane and the subsequent endocytosis and trafficking to the nucleus in order to achieve transient gene expression. Improvements made to polymers or lipids have resulted in numerous commercially available products for highly efficient transfection. Methods to improve a cell’s ability to take in plasmid DNA have identified that transmembrane receptors can serve as ports of entry for the internalization of extracellular DNA. While there are thousands of receptors that can be present on a cell, the transferrin receptor 1 (TfR1) is ubiquitously expressed, as every cell requires extracellular iron for health and growth.
Given the success of transferrin as a molecular carrier, the use of transferrin has still relied on serum-derived transferrin from either bovine serum or human serum. Both of which are subject to numerous disadvantageous factors such as adventitious agents present in serum, constraints on the supply-chain, or increasing cost due to demand. While excess transferrin in transfection has already been identified to increase transfection efficiency, we show that blood-free and serum-free transferrin added in excess during the transfection of adherent cells improves the transfection efficiency compared to low levels of transferrin and serum-derived transferrin. We also show that the inclusion of transferrin in transient transfection for viral production enhanced functional viral titers for lentivirus.
Materials and Methods
Cell Culture Conditions
HEK-293T (293T, ATCC CRL-3216) and HT-1080 (ATCC CCL-121) were maintained in either DMEM + 10% FBS for serum conditions, or complete OptiPEAK HEK293t® (InVitria, Junction City, Kansas), for serum-free conditions. Serum cultured cells were grown in standard conditions (5% CO2, 37 °C) while serum-free cells were grown in 10% CO2 and 37 °C. Cells were adapted to serum-free conditions by direct adaptation from FBS passaged cells. Both cell types were maintained by passaging with TrypLE (ThermoFisher #A1217701) every 3-4 days and seeded at initial densities of 10,000 cells/cm2.
Transfection Procedures
293T cells for transfection were plated 24 hours prior to transfection at a density of 50,000 cells per cm2 for GFP efficiency assays or for lentivirus production cells were plated at a density of 75,000 cells per cm2. For transfection efficiency with a GFP reporter, the plasmid Monster Green® Fluorescent Protein phMGFP Vector (Promega, Madison, WI) was used at a concentration of 0.2 µg/cm2. Lentivirus plasmids were delivered at a concentration of 0.16 µg/cm2. In all cases, PEIpro (Polyplus, Alsace, France) was used at a ratio of 1:1 with plasmid DNA for formation of DNA complexes.
Transferrin supplementation was carried out by adding reconstituted Optiferrin® (InVitria, Junction City, Kansas) to complete OptiPEAK HEK293t at a final concentration of 1 mg/mL. To form complexes, plasmid DNA and PEIpro were first diluted separately 40-fold. Diluted DNA was then added to diluted PEIpro, the contents were mixed by gentle pipetting or swirling, and complexes were formed under sterile conditions for 15-20 minutes at room temperature. Cells were incubated in normal growth conditions for 2.5 hours ± 30 minutes, after which transfection complexes were aspirated off of cells, and normal growth media were added back to cells (without additional transferrin for transferrin enhancement).
Viral Production and Titration
HT-1080 cells were used for functional transduction assays and were plated at a density of 30,000 cells per well in 12 well plates two hours before the assay. Lentivirus viral particles were harvested by collecting the total supernatant 48 hours post-transfection.
Results
Transfection Efficiency with GFP Reporter
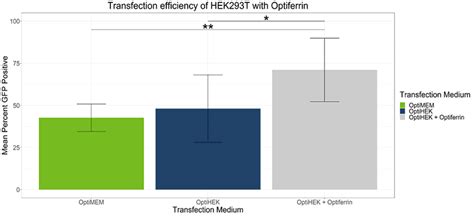
In standard conditions, OptiPEAK HEK293t achieves equivalent transfection efficiency compared to transfections performed under serum conditions complexed with OptiMEM. The average percentage of GFP positive cells complexed with OptiMEM and grown in DMEM + 10% FBS was 42.6% ± 8.1% and the average with OptiPEAK HEK293t was 48.1% ± 20.1% (Figure 1, difference in means is not statistically significant).
Enhancement of Functional Lentivirus Titer
To determine if transferrin enhancement of transfection efficiency correlated with higher viral titers, we performed transient transfection with lentivirus plasmids for viral production with 293T cells. Excess transferrin in the transfection step for viral production led to an increase in functional viral titer for lentivirus compared to Complete OptiPEAK HEK293t without excess transferrin (Figure 2).
Lentivirus production showed similar trends to transfection efficiency assays with a GFP reporter. Second generation lentivirus vectors were produced using a three plasmid system, and total lentivirus vectors were harvested 48 hours post transfection.
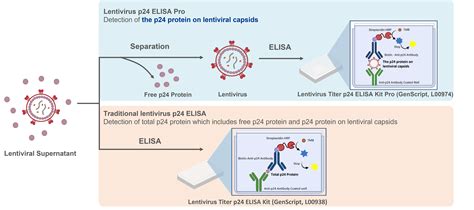
Plasmids complexed with OptiPEAK HEK293t and grown in chemically defined conditions with OptiPEAK HEK293t yielded average titer of 2.3 ± 1.9 × 107 IFU/mL (Figure 2, difference in means is not statistically significant). Together, these results demonstrate that excess transferrin does not inhibit DNA complexation and in fact improves functional lentivirus titer compared to chemically defined conditions without excess transferrin and improves functional titer compared to serum-derived transferrin in OptiMEM.
The improvements shown here with excess transferrin suggest that 293T cells may be more rapidly endocytosing plasmid DNA from the complexation medium.
Factors Influencing Transfection Efficiency
Cell Type and Passage Number
The choice of which cell type to use for a transfection experiment may seem obvious, but it is a critical factor that is often overlooked. Since each cell type is likely to respond differently to a given transfection reagent or method, choosing the appropriate cell type and proper experimental design are necessary to maximize results. While established continuous cell lines are easier to work with in the laboratory, they may not be the best choice for modeling in vivo processes because of the multiple genetic changes that they have undergone. However, if the purpose of the transfection experiment is high-level production of recombinant proteins, it is not important that the cell line represents the in vivo situation as long as the cell line can express sufficient quantities of recombinant proteins with proper folding and post-translational modifications.
For example, transient transfection of suspension-adapted Gibco Expi293F cells grown in Gibco Expi293 Expression Medium enables researchers to produce, starting from the vector of interest, greater than 1 g/L of correctly folded and glycosylated recombinant proteins. Primary cultures, on the other hand, are often used because they more closely mimic natural tissues. However, they typically have a limited growth potential and life span, and are more difficult to maintain in culture. When using primary cultures, it is important to maintain a largely homogeneous population of cells (for example, neuronal cultures should be enriched for neurons and suppressed with regard to glial cells) and use the cells as soon as practical.
Cell cultures with immortalized cell lines evolve over months and years in the laboratory, resulting in changes in cell behavior with regard to transfection. Excessive passaging is likely to detrimentally affect transfection efficiency as well as total transgene expression level from the cell population as a whole. In general, we recommend using cells that have undergone less than 30 passages after thawing of a stock culture. Thawing a fresh vial of frozen cells and establishing low-passage cultures for transfection experiments allow the recovery of transfection activity. For optimal reproducibility, aliquots of cells of a low passage number can be stored frozen and thawed as needed. Allow 3 or 4 passages after thawing a new vial of cells.
Cell Density and Health
The viability and general health of cells prior to transfection is known to be an important source of variability from one transfection to another. In general, cells should be at least 90% viable prior to transfection and have had sufficient time to recover from passaging. We strongly recommend subculturing cells at least 24 hours before transfection to ensure that they recover from the subculture procedure and are in optimum physiological condition for transfection.
The optimal cell density for transfection varies for different cell types, applications, and transfection technology, and should be determined for every new cell line to be transfected. Maintaining a standard seeding protocol from experiment to experiment ensures that optimal confluency at the time of transfection is reliably achieved. With cationic lipid-mediated transfection, generally 70-90% confluency for adherent cells or 5 × 105 to 2 × 106 cells/mL for suspension cells at the time of transfection provides good results.
Make sure that the cells are not confluent or in stationary phase at the time of transfection, because actively dividing cells take up foreign nucleic acid better than quiescent cells. Too high of a cell density can cause contact inhibition, resulting in poor uptake of nucleic acids and/or decreased expression of the transfected gene. However, too few cells in culture may result in poor growth without cell-to-cell contact. In such cases, increasing the number of cells in culture improves the transfection efficiency. Similarly, actively dividing cell lines are more efficiently transduced with viral vectors.
Culture Medium and Serum
Different cells or cell types have very specific medium, serum, and supplement requirements, and choosing the most suitable medium for the cell type and transfection method plays a very important role in transfection experiments. Information for selecting the appropriate medium for a given cell type and transfection method is usually available in published literature, and may also be obtained from the source of the cells or cell banks. If there is no information available on the appropriate medium for your cell type, you must determine it empirically.
It is important to use fresh medium, especially if any of the components are unstable, because medium that is missing key components and necessary supplements may harm cell growth.
In general, the presence of serum in culture medium enhances transfection with DNA. However, when performing cationic lipid-mediated transfection, it is important to form DNA-lipid complexes in the absence of serum because some serum proteins interfere with complex formation. Note that the optimal amounts of cationic lipid reagent and DNA may change in the presence of serum; thus, transfection conditions should be optimized when using serum-containing transfection medium.
When transfecting cells with RNA, we recommend performing the transfection procedure in the absence of serum to avoid possible contamination with RNases. Most cells remain healthy for several hours in a serum-free medium.
The quality of serum can significantly affect cell growth and transfection result. Therefore, it is important to control for variability among different brands or even different lots of serum to obtain best results. After testing the serum on your cells, keep using the same serum to avoid variation in your result.
Antibiotics and Nucleic Acid Quality
In general, antibiotics can be present in the medium for transient transfection. However, because cationic lipid reagents increase cell permeability, they may also increase the amount of antibiotics delivered into the cells, resulting in cytotoxicity and lower transfection efficiency. Therefore, we do not recommend adding antibiotics to the transfection medium. Avoiding antibiotics when plating cells for transfection also reduces the need for rinsing the cells before transfection.
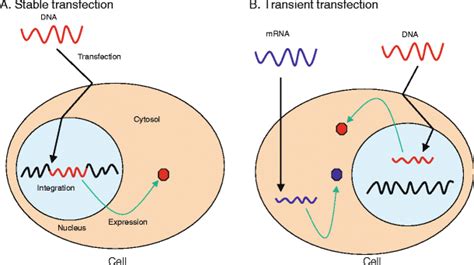
For stable transfections, penicillin and streptomycin should not be used in selective medium, because these antibiotics are competitive inhibitors of the Gibco Geneticin selective antibiotic.
Plasmid DNA is the most commonly used vector for transfection. The topology (linear or supercoiled) and the size of the plasmid DNA vector influence the efficiency of transfection. Transient transfection is most efficient with supercoiled plasmid DNA.
Transfection Reagents and Incubation Time
There are a number of strategies for introducing nucleic acids into cells that use various biological, chemical, and physical methods. However, not all of these methods can be applied to all types of cells and experimental applications, and there is a wide variation with respect to transfection efficiency, cell toxicity, effects on normal physiology, level of gene expression etc. The ideal approach should be selected depending your cell type and experimental needs, and should have high transfection efficiency, low cell toxicity, minimal effects on normal physiology, and be easy to use and reproducible.
Many chemical transfection reagents have an ideal time window, in which a transfection complex of optimal diameter is formed. This is typically between 5 and 30 minutes, depending upon the nature of the reagent. In general, transfection reagents need to be in contact with cells for a period of time before additional medium is added or the medium is replaced (to help minimize toxic effects of the reagent). In some instances, plating cells onto wells or plates containing transfection complexes may result in increased transfection efficiency, compared to the traditional approach of adding transfection complexes to an established culture. An additional benefit to such reverse transfection protocols is that seeding and transfecting cells on the same day shortens the experimental timeline by a full day.
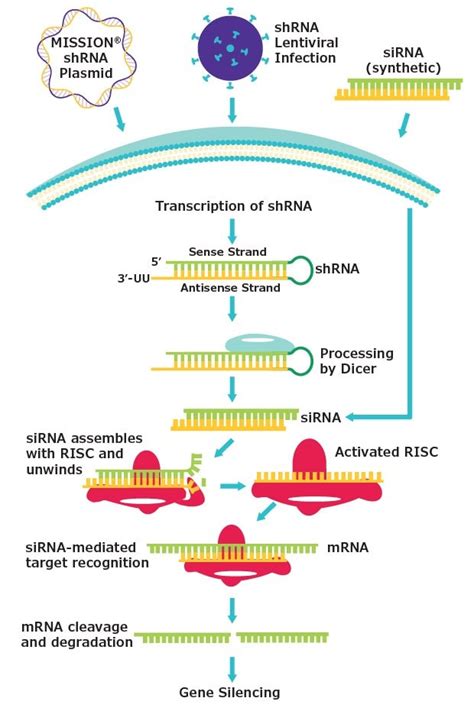
siRNA Transfection Protocol
Considerations for Specific Cell Types and Applications
Difficult-to-Transfect Cells
Some cell types, such as MCF7 or HepG2, prefer to grow in clumps or clusters which is not ideal for transfection because minimal membrane surface is exposed which compromises uptake; there are other cell types, such as blood or immune cells, that lack the proper endocytic machinery, which again can minimize uptake; and there are other cells, such as macrophages, that have an evolved uptake mechanism, but quickly breakdown and destroy endosomal contents.
Do you really need to use this cell type? These notoriously difficult cells require a certain amount preparation, because a variety of delivery techniques might need to be tested for success. If you are trying to over express a gene with DNA delivery, the newly developed Lipofectamine® 3000 can be tried first for the simplicity of using a lipid based delivery method. You could also try to do electroporation with an mRNA transcript of your gene of interest, which only needs to be delivered to the cytoplasm for protein translation. If all these previous methods fail, a virus can be used for gene expression.
Primary Cells and mRNA Delivery
For delivery in primary cells or any cell model, gene expression is dependent on the downstream assay required for the experiment. A typical window or range is anywhere from as early as 12 hours for expression from mRNA delivery, 24-48 hours for evaluation of expression of a fluorescent protein, to 72-96 hours for evaluation of genome editing tools like TALs or CRISPR. However, there are factors that could influence the efficiency of expression such as size of the DNA or mRNA, the promoter for the DNA, age and health of the cells, the half-life of the protein being expressed, etc.
On another note, a little tip we can offer, if you are trying to improve delivery in primary cells, would be to try an mRNA version of your gene of interest. The reasoning is that mRNA only requires entry to the cytoplasm of the cell as opposed to the nuclear localization that is required for DNA; and since primary cells tend to be in a post-mitotic state, with minimal nuclear envelope disruption, delivery to the nucleus has proven to be quite challenging. Life Technologies offers multiple in vitro transcription kits that are complete with the necessary items to make mRNA in the lab; an ARCA cap, the nucleotides and a RNA specific polymerase are included in the kit. The only requirement is a linearized vector or PCR product of the gene of interest containing a T7 or SP6 promoter.
Co-transfection of DNA and siRNA
I am trying to co-transfect DNA and siRNA into HEK cells. For getting successful co-transfection, you need high quality DNA, a validated siRNA against the gene of interest and log phase growing HEK293 cells. To study the effect of siRNA knockdown of an endogenous gene, siRNA transfection should be done first using an RNAi specific delivery reagent such as Lipofectamine® RNAiMax. 4-48 hours after siRNA has been delivered, DNA can be transfected using Lipofectamine® 3000 or Lipofectamine® 2000. Once the appropriate method of delivery is determined based on the application, transfection should be optimized for DNA, siRNA and lipid delivery reagent doses.
Troubleshooting Common Transfection Issues
Drop in Cell Viability
After transfection, I regularly get a drop in cell viability. You might try removing the media that contains the transfection complex 4-6 hours post transfection and replace it with growing culture media. If this doesn’t help with cell viability, you might need to do a small experiment to optimize a few key parameters that can influence the performance of your transfection.
Cell density can be an important factor for cell viability and transfection. If cell density is too low at the time of transfection, then a big drop in viability may be seen. Also, ensure that high quality DNA is used.
Inconsistent Results
Successful transfection is influenced by many factors-the choice of the transfection method, health and viability of the cell line, number of passages, degree of confluency, quality and quantity of the nucleic acid used, and the presence or absence of serum in the medium can all play a part in the outcome of your transfection experiment.
For optimal reproducibility, aliquots of cells of a low passage number can be stored frozen and thawed as needed. Allow 3 or 4 passages after thawing a new vial of cells.
Since contamination can drastically alter transfection results, cell cultures and media should be routinely tested for biological contamination (see Biological Contamination), and contaminated cultures and media should never be used for transfection.
The optimal cell density for transfection varies for different cell types, applications, and transfection technology, and should be determined for every new cell line to be transfected. Maintaining a standard seeding protocol from experiment to experiment ensures that optimal confluency at the time of transfection is reliably achieved.
Similarly, actively dividing cell lines are more efficiently transduced with viral vectors. Different cells or cell types have very specific medium, serum, and supplement requirements, and choosing the most suitable medium for the cell type and transfection method plays a very important role in transfection experiments. Information for selecting the appropriate medium for a given cell type and transfection method is usually available in published literature, and may also be obtained from the source of the cells or cell banks. If there is no information available on the appropriate medium for your cell type, you must determine it empirically.
It is important to use fresh medium, especially if any of the components are unstable, because medium that is missing key components and necessary supplements may harm cell growth.
tags: #serum #in #culture #media #transfection #performance